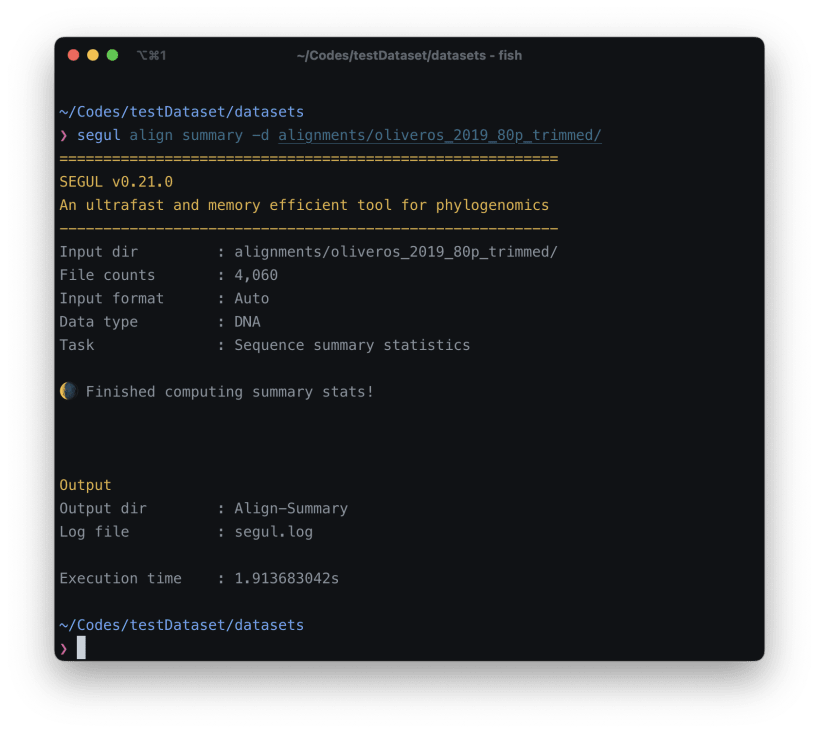

SEGUL is an ultrafast, memory efficient, cross-platform tool for phylogenomic data manipulation and summary statistics calculation. We develop SEGUL to address the need for a high-performance and accessible phylogenomic tool. It is particularly well-suited for large-scale phylogenomic projects, especially those involving thousands of loci and hundreds of samples. Additionally, SEGUL is capable of handling small Sanger sequences effectively.
Features:
You are about to open
Do you wish to proceed?
Thank you for your report. Information you provided will help us investigate further.
There was an error while sending your report. Please try again later.
Snaps are applications packaged with all their dependencies to run on all popular Linux distributions from a single build. They update automatically and roll back gracefully.
Snaps are discoverable and installable from the Snap Store, an app store with an audience of millions.
If you’re running Kubuntu 16.04 LTS (Xenial Xerus) or later, including Kubuntu 18.04 LTS (Bionic Beaver) and Kubuntu 18.10 (Cosmic Cuttlefish), you don’t need to do anything. Snap is already installed and ready to go.
Versions of Kubuntu between 14.04 LTS (Trusty Tahr) and 15.10 (Wily Werewolf) don’t include snap by default, but snap can be installed from the command line as follows:
sudo apt update
sudo apt install snapd
To install segul, simply use the following command:
sudo snap install segul
Browse and find snaps from the convenience of your desktop using the snap store snap.

Interested to find out more about snaps? Want to publish your own application? Visit snapcraft.io now.

